
- Criteria #1
- Criteria #2
- Criteria #3
- Criteria #4
- Criteria #5
-
Characteristics
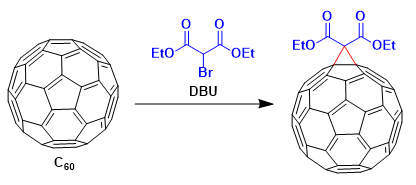
The Bingel reaction in fullerene chemistry is a fullerene cyclopropanation reaction to a methanofullerene first discovered by C. Bingel in 1993[a] with the bromo derivative of diethyl malonate in the presence of a base such as sodium hydride or DBU(1,8-Diazabicyclo[5.4.0]undec-7-ene). This reaction is a most popular tool for the functionalization of C60 derivatives. Alternative strategies to generate reactive monohalomalonate intermediate in situ have been reported by Hirsch[b]
The advantages of this method are: (i) exclusive addition on [6,6] double bonds of the fullerene skeleton, (ii) mild reaction conditions giving relatively high yields, (iii) installation of ester moieties that allow additional chemical transformations.
-
Literature reference
[a] Bingel, C. Chem. Ber. 1993, 126, 1957. DOI:10.1002/cber.19931260829
[b] Hirsch, A.; Camps, X. J. Chem. Soc., Perkin Trans. 1, 1997, 1595. DOI: 10.1039/A702055D
-
Reaction mechanism
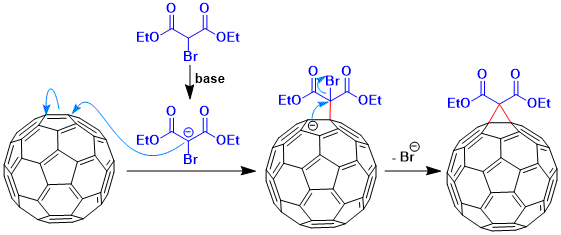
The alpha-halomalonate anion is added to C60 and this is followed by intramolecular displacement of the halide by the anionic center generated on the fullerene core to give the corresponding cyclopropanated [60]fullerene.
-
Example of reactions
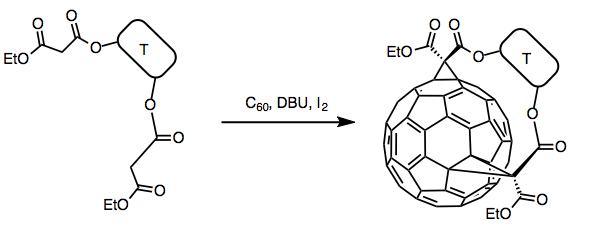
In 1994, a general approach for the tether-directed double Bingel cyclopropanation was proposed by Diederich and coworkers[1]
This approach involves reaction of C60 with a tether-connected bismalonate in the presence of I2 and DBU. The corresponding 2-iodo-malonate was generated in situ and subsequently reacted with C60. Tether-directed remote functionalization gave improved yields and regioselectivity.
Bingel reaction can be applied to the functionalization of metallofullerenes.[2]
-
Bibliography
[1] “Spacer-Controlled Multiple Functionalization of Fullerenes”
Thilgen, C.; Sergeyev. S.; Diederich, F. Topics in Current Chemistry, 2004, 248, 1.
The chapter provides a survey of the development and applications of the tether- and template-directed regio- and, in the occurrence, stereoselective multifunctionalization of fullerenes over the past ten years. After a presentation of the first tether-directed remote functionalization of C60, a broad spectrum of applications is reviewed according to the involved reaction types. The most frequently used chemistry consists of additions of tethered 2-halomalonates (double Bingel reactions) and 1,3-dienes (double Diels-Alder reactions). The former, in particular, were used for the only known tether-directed functionalization of a higher fullerene (C70) and also for most of the rare examples of three- and fourfold one-pot tether-directed derivatizations of C60. Other, commonly used reactions are [3+2] cycloadditions, notably of azides and, in a few cases, of vinylcarbenes, and azomethine ylides. Some interesting examples of intramolecular [2+2] cycloadditions between fullerene moieties are also included as they are in fact spacer-controlled dimerizations of the carbon spheres. Throughout the account, particular emphasis is put on the diastereoselective generation of chiral fullerene functionalization patterns by use of enantiomerically pure tethers. As compared to tether-directed multiple additions to fullerenes, regioselective functionalizations with non-covalent templates have remained rather rare. Two important examples are reported, one involving a reversible reaction with 9,10-dimethylanthracene in solution, the other one a topochemical anthracene transfer.
[2] “Bingel−Hirsch Reactions on Non-IPR Gd3N@C2n (2n = 82 and 84)”
Alegret, N.; Chaur, M. N.; Santos, E.; Rodríguez-Fortea, A.; Echegoyen, L.; Poblet, J. M. J. Org. Chem. 2010, 75, 8299–8302. DOI: 10.1021/jo101620b
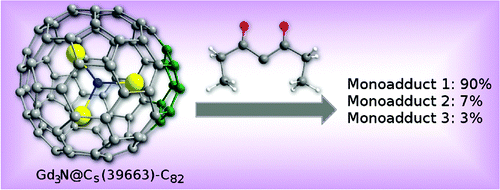
The Bingel−Hirsch reactions on non-isolated pentagon rule (non-IPR) Gd3N@C2n (2n = 82, 84) are studied. Computational results show that the two metallofullerenes display similar reactivity according to their related topologies. Long C−C bonds with large pyramidalization angles lead to the most stable adducts, the [5,6] bonds in the adjacent pentagon pair being especially favored. The lesser regioselectivity observed for Gd3N@C82 is probably due to the activation of some C−C bonds by means of the metal cluster.
-
Related Books
[amazonjs asin=”3642083811″ locale=”US” title=”Carbon Rich Compounds I (Topics in Current Chemistry)”]
-
Related Links
Bingel Reaction - Wikipedia