
DiRocco, D. A.; Dykstra, K.; Krska, S.; Vachal, P.; Conway, D. V.; Tudge, M. Angew. Chem. Int. Ed. 2014, Early View.
DOI: 10.1002/anie.201402023
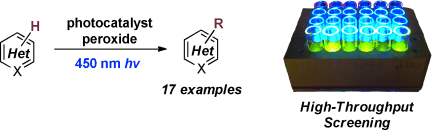
The direct C–H functionalization of heterocycles has become an increasingly valuable tool in modern drug discovery. However, the introduction of small alkyl groups, such as methyl, by this method has not been realized in the context of complex molecule synthesis since existing methods rely on the use of strong oxidants and elevated temperatures to generate the requisite radical species. Herein, we report the use of stable organic peroxides activated by visible-light photo- redox catalysis to achieve the direct methyl-, ethyl-, and cyclopropylation of a variety of biologically active hetero- cycles. The simple protocol, mild reaction conditions, and unique tolerability of this method make it an important tool for drug discovery.
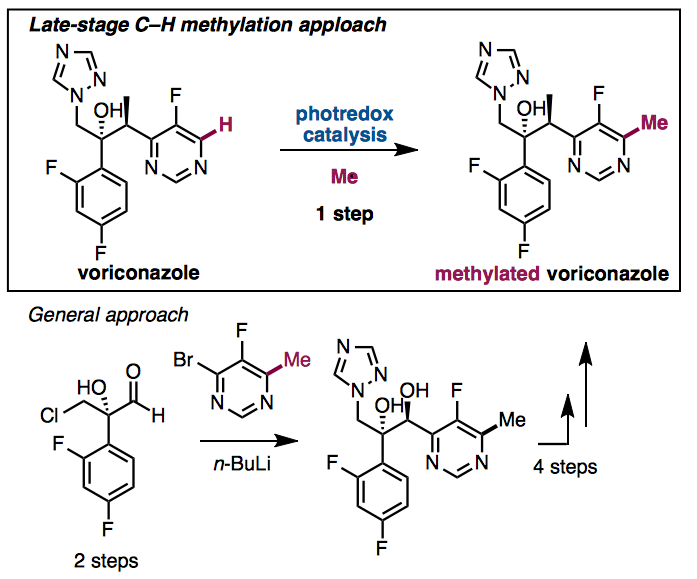
Late stage C–H functionalization has attracted much attention for the-state of art synthetic tool on organic chemistry. DiRocco and coworkers demonstrated late-stage C–H methylation[1] of N-heteroaromatics in this paper by using photoredox catalysis. They used High-throughput experimentation which developed by MacMillan et al for discovering the best catalyst. [2] They applied their reaction to functionalized molecules, for examples voriconazole, clinically used triazole anti fungal agent. Shibasaki group have already accomplished the efficient synthesis of voriconazole in 7 steps using catalytic asymmetric cyanosilylation as a key step.[3] However, if you would like to put methyl on the compounds, we have to prepare it at the initial stage on the synthesis. On the other hand, you can be introduce the methyl group directly from voriconazole by using their developed photredox catalyst system.
-
References
[1] Another example, “C−H Methylation of Heteroarenes Inspired by Radical SAM Methyl Transferase”
Gui, J.; Zhou, Q.; Pan, C.-M.; Yabe, Y.; Burns, A. C.; Collins, M. R.; Ornelas, M. A.; Ishihara, Y.; Baran, P. S. J. Am. Chem. Soc. 2014, 136, 4853. DOI: 10.1021/ja5007838
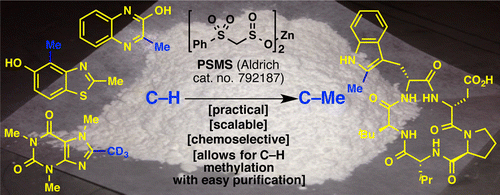
A practical C–H functionalization method for the methylation of heteroarenes is presented. Inspiration from Nature’s methylating agent, S-adenosylmethionine (SAM), allowed for the design and development of zinc bis(phenylsulfonylmethanesulfinate), or PSMS. The action of PSMS on a heteroarene generates a (phenylsulfonyl)methylated intermediate that can be easily separated from unreacted starting material. This intermediate can then be desulfonylated to the methylated product or elaborated to a deuteriomethylated product, and can divergently access medicinally important motifs. This mild, operationally simple protocol that can be conducted in open air at room temperature is compatible with sensitive functional groups for the late-stage functionalization of pharmacologically relevant substrates.
[1] “Discovery of an α-Amino C–H Arylation Reaction Using the Strategy of Accelerated Serendipity”
McNally, A.; Prier, C. K.; MacMillan, D. W. C. Science 2011, 334, 1114. DOI: 10.1126/science.1213920
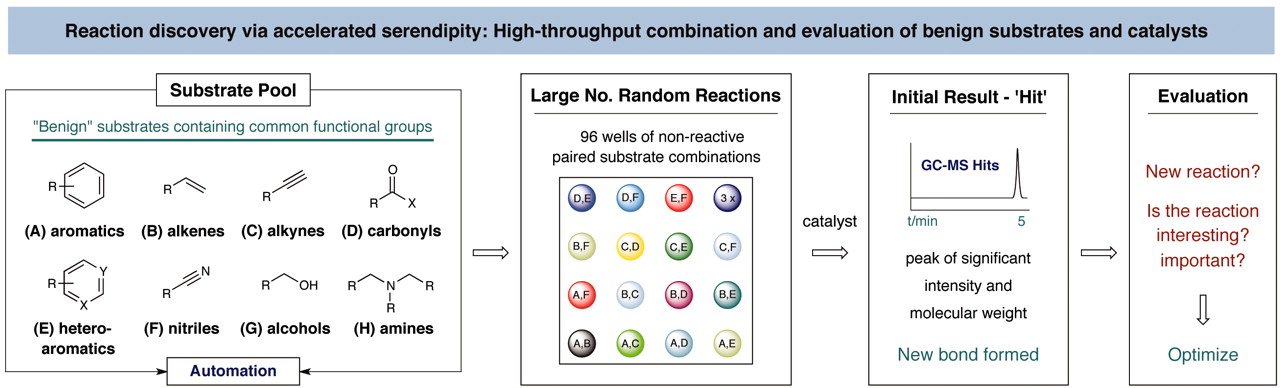
Serendipity has long been a welcome yet elusive phenomenon in the advancement of chemistry. We sought to exploit serendipity as a means of rapidly identifying unanticipated chemical transformations. By using a high-throughput, automated workflow and evaluating a large number of random reactions, we have discovered a photoredox-catalyzed C–H arylation reaction for the construction of benzylic amines, an important structural motif within pharmaceutical compounds that is not readily accessed via simple substrates. The mechanism directly couples tertiary amines with cyanoaromatics by using mild and operationally trivial conditions.
[1] “An Enantioselective Synthesis of Voriconazole”
Tamura, K.; Furutachi, M.; Kumagai, N.; Shibasaki, M. J. Org. Chem. 2013, 78, 11396. DOI: 10.1021/jo4019528
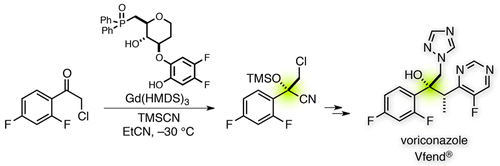
A new seven-step sequence to access voriconazole, a clinically used antifungal agent, was developed. The initial catalytic asymmetric cyanosilylation is the key to constructing the consecutive tetra- and trisubstituted stereogenic centers. The fluoropyrimidine unit frequently triggered unexpected side reactions, but careful amendment of the reaction sequence allowed for the concise enantioselective synthesis.
-
Related Books
[amazonjs asin=”0841238499″ locale=”US” title=”Activation and Functionalization of C-H Bonds (Acs Symposium Series, 885)”]