

Kohei Tamao (Born in 1942) is an organic chemist in Japan. He is currently the Chemical Society of Japan.
Kohei Tamao received his B.S. (1965), and Master’s (1967) degrees from Kyoto University (Dept. of Synthetic Chemistry, Faculty of Engineering). He completed his doctoral course in 1970 and was awarded a Doctor of Engineering in 1971. After a postdoctoral position at the State Univ. of New York at Binghamton with J.J. Eisch, he joined Kyoto University as an Assistant Professor. He was promoted to Associate Professor in 1987 and then to Professor in 1993. In 2000, he was appointed Director the Institute for Chemical Research at Kyoto University. This appointment was followed by an appointment as Director of the International Research Center for Elements Science. In 2005, he was awarded the title of Professor Emeritus at Kyoto University. He is the present Director of the RIKEN Frontier Research System.
- Education and Experience
1965 B.S. Kyoto University, Dept. of Synthetic Chemistry, Faculty of Engineering
1967 M.S. Kyoto University, Dept. of Synthetic Chemistry, Faculty of Engineering
1970 Assistant Professor, Kyoto University
1971 D.Eng., Kyoto University
1993 Professor, Institute for Chemical Research, Kyoto University
2000 Director, Institute for Chemical Research, Kyoto University
2005 Director, RIKEN Frontier Research System
2006 Unit Leader, Functional Elemento-Organic Chemistry Unit, RIKEN
2008 Director, RIKEN Advanced Science Institute
- Awards and Honors
1977 Chemical Society of Japan Award for Young Chemists
1999 Chemical Society of Japan Award
2002 Frederic Stanley Kipping Award of the American Chemical Society
2002 Toray Science & Technology Prize
2003 Asahi Prize (Asahi News Paper Culture Foundation, 2003)
2004 Medal of Honor with Purple Ribbon
2007 Japan Academy Prize
2011 A Person of Cultural Merits
- Research
Prof. Tamao is responsible for introducing a new concept in bond activation involving high coordination in modern synthetic organic chemistry, by developing a variety of bond cleavage reactions among hexacoordinate organosilicon compounds. Based on these developments, he discovered hydrogen peroxide oxidation of silicon-carbon bonds in ordinary tetracoordinate organosilicon compounds by introducing electronegative group(s) on silicon as essential substituents for the formation of activated hyper coordinate species.
The Kumada-Tamao-Corriu coupling is a type of cross coupling reaction, useful for generating carbon–carbon bonds by the reaction of a Grignard reagent and an organic halide. The procedure uses transition metal catalysts, typically nickel or palladium, to couple combination of two alkyl, aryl or vinyl groups. When Prof. Tamao was an assistant professor in Kumada group, the groups of Robert Corriu and Makoto Kumada (with Kohei Tamao) reported the reaction independently in 1972.[1]
![]()
This oxidation reaction, now known as Tamao-Fleming oxidation, represents the only existing general method for the synthesis of alcohols from organosilicon compounds, and has overturned the previously accepted wisdom among organic chemists that silicon-carbon bonds are fairly resistant to oxidative cleavage, greatly enhancing the synthetic utility of organosilicon compounds.[2]
![]()
Another new chemical development in main-group element compounds developed by him is the control of photophysical properties by coordination number change. A dramatic color change and a fluorescence change in boron and silicon compounds having three anthracene rings have been demonstrated through the addition of a fluoride ion which causes the coordination number change, further providing basic ideas for fluoride ion sensors.
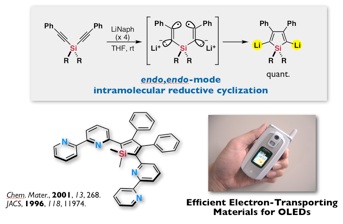
He has also demonstrated that silole, a silicon-containing five-membered cyclic compound, is an electron-accepting molecule due to a unique orbital interaction involving a silicon atom.[3] He developed a new general synthetic method for the introduction of silole rings into a variety of π -conjugated systems and applied, through collaboration with corporate interests, 2,5-dipyridyl-silole derivatives to electroluminescent (EL) devices as the most efficient electron-transporting materials, and which have achieved practical application in the production of small, full-color displays on commercial mobile telephones.
- Representative Papers
[1] (a) Tamao, K.; Sumitani, K.; Kumada, M. J. Am. Chem. Soc. 1972, 94, 4374. DOI:10.1021/ja00767a075 (b) Corriu, R. J. P.; Massse, J. P. J. Chem. Soc., Chem. Commun. 1972, 144. DOI: 10.1039/C3972000144a
[2] Tamao, K.; Akita, M.; Kumada, M. Organometallics 1983, 2, 1694. DOI:10.1021/om50005a041
[3] Tamao, K.; Uchida, M.; Izumizawa, T.; Furukawa, K.; Yamaguchi, S. J. Am. Chem. Soc. 1996,118, 11974. DOI: 10.1021/ja962829c
[4] A Planar Rhombic Charge-Separated Tetrasilacyclobutadiene
Suzuki, K.; Matsuo, T.; Hashizume, D.; Fueno, H.; Tanaka, K.; Tamao, K. Science 2011, 331, 1306–1309. DOI: 10.1126/science.1199906
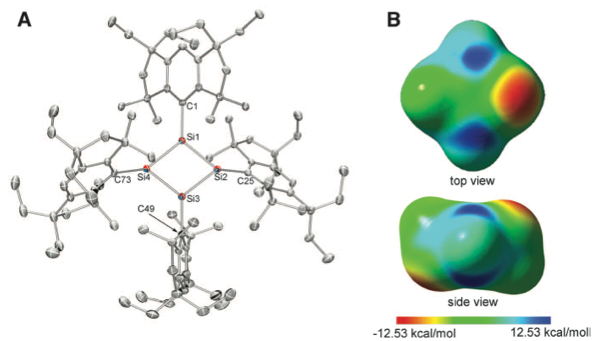
The cyclobutadiene (CBD) molecule C4H4 deviates from a high-symmetry square geometry to compensate for its antiaromatic electronic structure. Here, we report a CBD silicon analog, Si4(EMind)4 (1), stabilized by the bulky 1,1,7,7-tetraethyl-3,3,5,5-tetramethyl-s-hydrindacen-4-yl (EMind) groups, obtained as air- and moisture-sensitive orange crystals by the reduction of (EMind)SiBr3 with three equivalents of lithium naphthalenide. X-ray crystallography reveals a planar and rhombic structure of the Si4 four-membered ring, with alternating pyramidal and planar configurations at the silicon atoms. The large 29Si chemical shift differences (Δδ > 350 parts per million) in the solid-state nuclear magnetic resonance spectra suggest a contribution of an alternately charge-separated structure. The rhombic-shaped charge-separated singlet state of compound 1 thus stabilizes its cyclic 4π-electron antiaromaticity in a manner that contrasts sharply with the bond-length alternation, characterizing the rectangular distortion of carbon-based CBD.
[5] A stable germanone as the first isolated heavy ketone with a terminal oxygen atom
Li, L.; Fukawa, T.; Matsuo, T.; Hashizume, D.; Fueno, H.; Tanaka, K.; Tamao, K. Nat/ Chem. 2012, 4, 361–365. DOI: 10.1038/nchem.1305
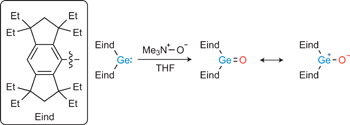
The carbon–oxygen double bond of ketones (R2C5O) makes them among the most important organic compounds, but their homologues, heavy ketones with an E5O double bond (E 5 Si, Ge, Sn or Pb), had not been isolated as stable compounds. Their unavailability as monomeric molecules is ascribed to their high tendency for intermolecular oligomerization or polymerization via opening of the E5O double bond. Can such an intermolecular process be inhibited by bulky protecting groups? We now report that it can, with the first isolation of a monomeric germanium ketone analogue (Eind)2Ge5O (Eind 5 1,1,3,3,5,5,7,7-octaethyl-s-hydrindacen-4-yl), stabilized by appropriately designed bulky Eind groups, with a planar tricoordinate germanium atom. Computational studies and chemical reactions suggest the Ge5O double bond is highly polarized with a contribution of a charge-separated form (Eind)2Ge12O2. The germanone thus exhibits unique reactivities that are not observed with ordinary ketones, including the spontaneous trapping of CO2 gas to provide a cyclic addition product.
- Photo Gallery







- Related Books
[amazonjs asin=”3527312145″ locale=”US” title=”Organosilicon Chemistry VI: From Molecules to Materials (2 Volumes)”][amazonjs asin=”1244571288″ locale=”JP” title=”Articles on Organoboron Compounds, Including: Borabenzene, Borazine, Borole, 1,2-Dihydro-1,2-Azaborine, Borirane, Bortezomib, Boronic Acid, Phenylboro”]