
An efficient catalyst that is cheap, air and moisture tolerant, with no toxicity….
The development of such a catalyst would merit, at the very least, candidacy for the Nobel prize in Chemistry. Over the years, numerous catalysts that utilize transition metal elements have found their place in industrial processes, and many of these exhibit desirable reactivities while maintaining high efficiency. A prominent drawback of these catalysts, however, is their toxicity. Since the turn of the new century organocatalysts, or catalysts composed of organic molecules, have been gaining great interest. These catalysts possess the advantage of low toxicity and air/moisture tolerance relative to those that utilize transition metals.
Why bother with transition metal catalysts then? The simple answer is their diverse reactivity. Because known organocatalysts are mainly used to facilitate so-called ionic reactions, whereby an electron donor/acceptor pair is involved, their reactivity is limited; perhaps inevitably so due to their only recent gain in interest. One of the most important studies in this field is to increase reactivity variation.
One conceivable path to this goal is to focus on non-ionic reactions; radical or pericyclic reactions, for example. In this post, I’d like to introduce an interesting asymmetric catalytic reaction developed by Prof. Keiji Marukoka’s group at Kyoto University. In this reaction, the organocatalyst takes the form of a sulfur radical (thiyl radical), opening the field of organocatalysts to non-ionic reactions.
“An organic thiyl radical catalyst for enantioselective cyclization”
Hashimoto, T.; Kawamata, Y, Maruoka, K. Nat. Chem. 2014, ASAP. DOI: 10.1038/nchem.1998
- Catalysis with a Thiyl Radical
Thiyl radicals are unstable species that generally exist in the form of its dimers (disulfides, RS-SR), which are stable and are found in numerous products; i.e. allyldisulfides in onions. In order to generate thiyl radicals, energy must be spent to break the RS-SR S-S bond. A useful way to do so is light; sunlight may be used, but mercury lamps are often used due to its higher energy emission.
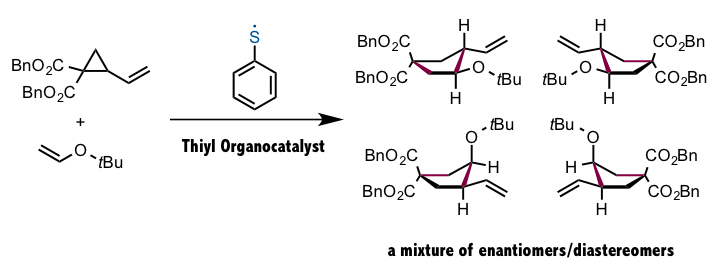
These thiyl radicals are precisely what Prof. Maruoka’s group has succeeded in using as catalysts for asymmetric catalysis. As it turns out, however, the use of thiyl radicals as organocatalysts in synthesis has a few precedents. Notably, Prof. Koichiro Oshima’s group in 1988 reported an extremely interesting radical cyclization reaction, as shown below. Unfortunately in this case, a mixture of enantiomers/diastereomers was found. Prof. Maruoka’s group designed a organocatalyst that could selectively generate a single stereoisomer, thereby building on the results of Prof. Oshima.
- Catalyst Design
This section will feature a slightly more technical discussion. The mechanism of the thiyl radical catalysis is shown below. The reaction starts with an attack onto the starting material double bond, and is followed by ring opening of the cyclopropane fragment, a second attack of the resulting radical onto the second starting material, and concludes with ring closing to yield the product. The chirality is thus dictated at the part of the reaction denoted by the grey arrow in the below figure, and any asymmetric catalyst for this reaction must exhibit selectivity at this step.
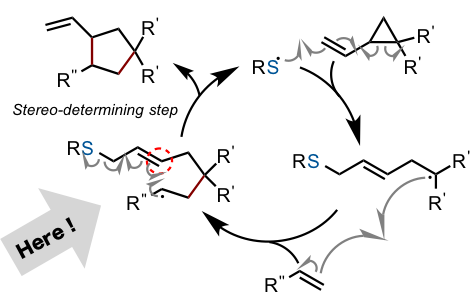
Reaction Mechanism
It was Prof. Takuya Hashimoto and his student Mr. Yu Kawamata, a first year graduate student at the time, in the Maruoka group who took upon the task of catalyst design. First they conceived of a thiyl radical catalyst that possessed a chiral naphthol (binaphthol-type) moiety. Initial radical generation was attempted via light treatment of the disulfide, but they eventually moved onto a different method that utilized benzoyl peroxide (BPO) and light to generate the thiyl radical from its corresponding thiol. After studying numerous binaphthol-type catalysts, they found that the best results were obtained by using a catalyst that possessed a Si(t-Bu)(2-Np)2 moiety as the SiAr2 unit in the red fragment shown in the below figure. This catalyst produced a 82% yield, with a 91:9 diastereoselectivity (44% ee). At this point they succeeded in selectively producing two of the four possible stereoisomers, with a 7:3 selectivity in the products.
After observing initial success with the binaphthol-type catalyst design, Mr. Kawamata subsequently made the bold move of using a more robust terphenyl moiety. While the change was based on theoretical considerations, such a move was indeed quite daring. Although we won’t go into the details, it’s fair to say the move paid off well, leading to a 95% yield, with a 95:5 diastereoselectivity (86% ee). Thus, Mr. Kawamata succeeded in producing nearly a single isomer. This incredible accomplishment was the result of three years of perseverance and hard work.
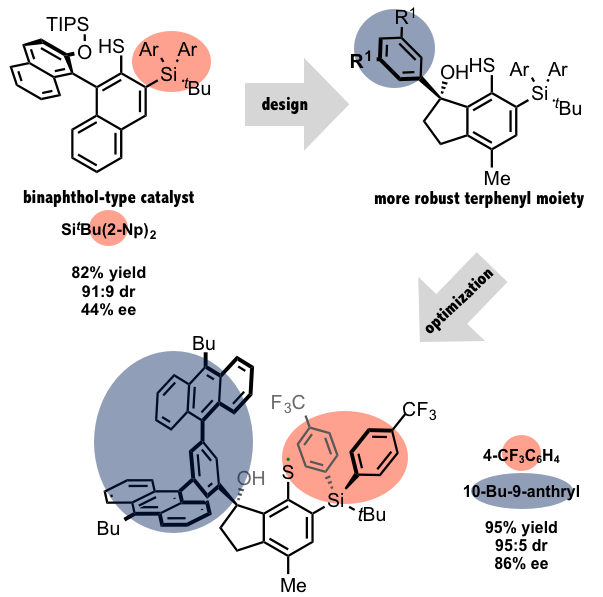
Catalyst Design
Although the thiyl radical organocatalyst requires 10 steps to make, the synthetic scheme is exquisite. Use of a Newman-Kwart rearrangement and a retro thia-Brook rearrangement allows for installment of the silyl group (shown below in red) at the appropriate location. Also noteworthy is that the aryl moiety is included towards the end of the synthesis.
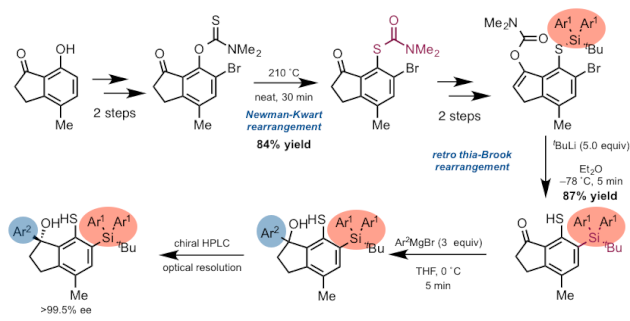
Synthetic Scheme
To summarize, this post highlighted a new organocatalyst that proceeds through a radical intermediate for asymmetric catalysis. This work is noteworthy in that a non-ionic reaction pathway is followed, despite the use of an organocatalyst, and hence expands on the reaction scope exhibited by these catalysts. We look forward to the subsequent studies, as applications towards other reactions and further improvements seem very plausible.
- Comments from the authors
Finally, we were fortunate enough to have the opportunity to interview the authors of this work.

{kind=link}
Prof Takuya Hashimoto
In the past, asymmetric catalysis by organocatalysts could be largely classified as catalysts with acid/base functionalities that facilitate ionic reactions. Members of the Maruoka group have been working hard to develop organocatalysts that exhibit reactivity that go beyond this general classification. Use of radical functionalities for controllable radical reactions is one such project. Towards this goal, we focused on Prof. Oshima and Prof. Feldman’s work in 1988 that described an asymmetric radical ring closing reaction by a thiyl radical.
Before officially launching this asymmetric organocatalysis project, I synthesized a simple binaphthol-type catalyst, which exhibited a rather lowly 5% ee. I wasn’t fully convinced that this would be worth pursuing, but I gave Mr. Kawamata the task of fulfilling our goal. Despite his extreme perseverance, we were initially unable to obtain desirable results, and oftentimes contemplated giving up. However, we reasoned based on the following logic/observations that our goal would be attainable;
A) The reaction does not proceed in the absence of catalyst
B) The transition state that leads to the stereoselectivity is clear
C) Nature uses thiyl radicals for stereoselective reactions.
At the same time, we also convinced ourselves that we’ve come too far to give up (over 2000 reactions). After long and countless hours, we became optimistic that we were within reach when we found that the product cis-trans ratio was nearly identical (Table1, Entry 5 in manuscript).
This research has shown us that enantioselective organocatalysis in radical reactions is possible. While we admit that the catalyst is rather complex, the radical ring closing reaction proceeds stereoselectively at 0 deg celcius and highlights its impressive reactivity. However, we’ve also observed that the selectivity is greatly dependent on the substrate, which is in contrast to the acid/base reactions we studied in the past. Another interesting aspect of this work was our approach of designing the catalyst from scratch to fit the needs of the reaction, not vice verca.
Mr. Yu Kawamata
I became involved in this project after a seemingly innocuous conversation with Prof. Hashimoto, in which he stated,
“We’d really like to use thiyl radicals in asymmetric ring closing organocatalysis. Here’s the substrate. Good Luck!”
From the start, we encountered countless difficulties in obtaining enantioselectivity in this reaction, not least of which were the intermediates with high degrees of freedom, chiral centers far separated from the catalyst, and radicals that were difficult to control. As perhaps expected, we were unable to find a viable catalyst. I attempted close to 100 binaphthol-type catalysts in a year, some of which required more than 10 steps to synthesize. After nearly running out of ideas of new variations, I came to a standstill.
It was around this time when I was able to obtain a crystal structure of a reaction intermediate using one of the best binaphthol-type catalysts at the time. The crystal structure revealed a silyl moiety that surrounded one half of the crucial sulfur atom, along with a naphthol moiety that seemed loosely associated with the chiral center. This structure provoked me to design a more robust chiral environment while maintaining the silyl moiety; in essence, a complete overhaul of the catalyst design.
It was not easy to give up on the binaphtol-type catalyst that I worked on for the past year, but luckily the change brought about good results. We used calculations to optimize the geometry of catalyst candidates, which led to the basic design in this work. We also made sure to install the silyl moiety and the aryl group near the chiral center towards the end of the synthesis to allow of ready optimization. This abridged synthetic strategy allowed for rapid screening of catalysts, which in turn led to our discovery. I near succumbed to this challenge on numerous occasions, but I feel great satisfaction in designing a new catalyst structure from theoretical studies, and to witness the catalyst perform at a high level.
- References
[1] Miura, K.; Fugami, K.; Oshima, K; Utimoto, K. Tetrahedron Lett. 1988, 29, 5135. DOI: 10.1016/S0040-4039(00)80701-7
- Related Links