
He, C.; Zhu, C.; Dai, Z.; Tseng, C.-C.; Ding, H. Angew. Chem. Int. Ed. 2013, 52, 13256–13260.
DOI: 10.1002/anie.201307426
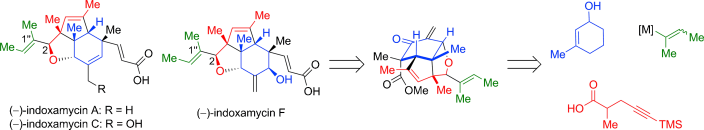
The concise and divergent total synthesis of (−)-indoxamycins A, C, and F has been completed for the first time by using a tricyclic enone as the common late-stage intermediate. The key steps of the strategy are based on an Ireland–Claisen rearrangement, a stereodivergent reductive 1,6-enyne cyclization, and a tandem 1,2-addition/oxa-Michael/methylenation reaction.
As a novel class of polyketides, indoxamycins A–F were isolated in 2009 by Sato et al. from saline cultures of marine- derived actinomycetes. The indoxamycin skeleton consists of an unprecedented [5,5,6] tricyclic cage-like carbon framework and two side chains having a trisubstituted olefin and an unsaturated carboxylic acid, respectively. In 2012, Carreira and co-workers reported an elegant total synthesis of rac-indoxamycin B ((1’’E)-2-epi-2), which led to a structural reassignment of the relative configuration at the C2 position and the geometry of the trisubstituted alkene in the side chain.[1]

The key steps of the strategy entail an Ireland–Claisen rearrangement, a stereo- divergent reductive 1,6-enyne cyclization, and a tandem 1,2- addition/oxa-Michael/methylenation reaction.
- References
[1] “Total Synthesis and Stereochemical Reassignment of (±)-Indoxamycin B”
Jeker, O. F.; Carreira, E. M. Angew. Chem. Int. Ed. 2012, 51, 3474–3477. DOI: 10.1002/anie.201109175
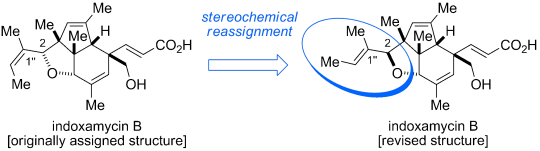
Revised version: The first total synthesis of indoxamycin B leads to a stereochemical reassignment of the natural product (see picture). The synthetic route features an efficient carboannulation sequence to rapidly access the dihydroindenone system. Moreover, a series of AuI-catalyzed transformations served in the construction of the sterically congested core framework.
- Related Links
Hanfeng Ding Research Group
- Related Books
[amazonjs asin=”3642340644″ locale=”US” title=”Total Synthesis of Natural Products: At the Frontiers of Organic Chemistry”][amazonjs asin=”3527329579″ locale=”US” title=”Classics in Total Synthesis III”]