
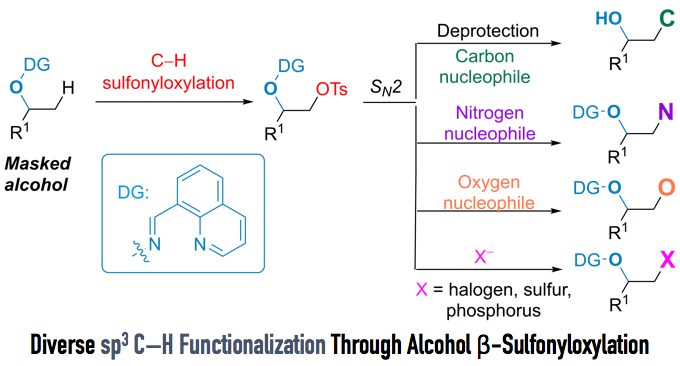
Sp3 C–H β-Sulfornyloxylation of Alcohols
Carbon–hydrogen bond activation (C–H activation) allows for the transformation of inherent C–H bonds within a molecule to various functional groups without pre-functionalization. In particular, transition metal-catalyzed C–H activation has become a major research area in present day organic synthesis. There has been a lot of C–H activations featured in this blog (Mainly for Japanese Chem-Station) as well. Recent related posts are as follows (including transition metal-free C–H activation).
- A meta-selective C–H borylation directed by a secondary interaction between ligand and substrate
- Sp3 C–H azidation by manganese catalyst
- Silylation of C–H bonds by alkali metal catalyst
- Hexaarylbenzenes
- Catalytic metal-free C–H borylation
- Convergent synthesis of (+)-Lithospermic Acid using C–H activation catalyst
Sp2 C–H activation has been achieved with relatively wide substrate generality; nevertheless sp3 C–H activation is much more difficult due to the inertness of the bond. Currently, there are many limitations in regioselectivity and functional group tolerance. It is essential to use a directing group (DG) – a “tag” that draws the transition metal catalyst closer through coordination – to activate the desired sp3 C–H bond. To date, numerous “tags” has been discovered in the research field of C–H bond activation, including those applicable to sp2 C–H activation.
Recently, Dong and coworkers at University of Texas in Austin reported the β-sp3 C–H sulfonyloxylation of alcohols by using a “tag” which they had developed. Tosylate can react with various nucleophiles for further transformations. In this article, we are pleased to introduce this work.
Y. Xu.; Guobing, Y.; Zhi, R.; Dong, G. “Diverse sp3 C−H Functionalization Through Alcohol β-Sulfonyloxylation”. Nature Chem. 2015, 7, 829-834. DOI: 10.1038/nchem.2326
Direct sp3 C–H Activation to Form Convenient Functional Groups (Figure 1)
The halogen is a typical reactive functional group that reacts with a variety of nucleophiles. Nucleophilic substitution of alkyl halides is one of the most common examples in the derivatization and functionalization of organic molecules. As written in your textbook, the conventional approach to sp3 C–H halogenation is mainly via radical halogenation. However, this process is plagued with side-reactions and poor regioselectivity. Moreover, radical halogenation is generally substrate-specific.
On the other hand, transition metal-catalyzed sp3 C–H activation has been developed in recent years and in particular, palladium-catalyzed regioselective sp3 C–H halogenation has been reported[1]. While DG-assisted β-halogenation of carbonyl proceeds in this reaction, deprotonation of acidic alpha-carbonyl proton potentially causes undesired elimination (or β-hydrogen elimination if transition metal is involved). This unproductive pathway produces olefins, removing the halogen that was painstakingly introduced. Thus, substrates are severely limited to the neopentyl group, which cannot undergo elimination (Figure 1).
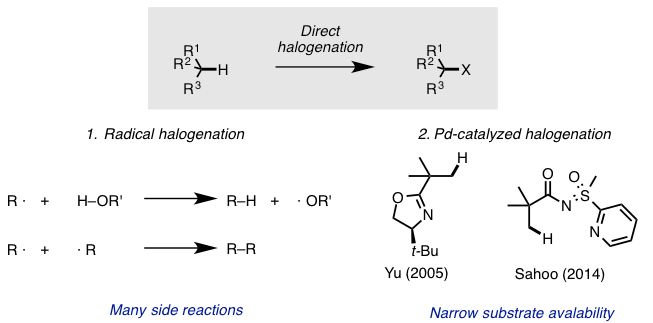
Figure 1. Current problem in C–H halogenation
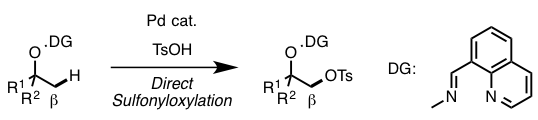
To overcome this drawback, Dong et al. attempted to achieve carbonyl-free direct sp3 β C–H functionalization of alcohols. They focused on the installation of sulfonyloxy group as a halogen equivalent.
-
Development of Direct Alcohol β-Sulfornyloxylation
The hint came from the authors’ previous work, catalytic sp3 C–H oxidation, which was reported in 2012[2]. Masking alcohol with oxime moiety as a directing group, the use of catalytic Pd(OAc)2 and oxidant PhI(OAc)2 under AcOH/Ac2O furnished β-acetoxylation[3]. Removal of N–O bond and acetyl group provides 1,2-diol (Figure 2).
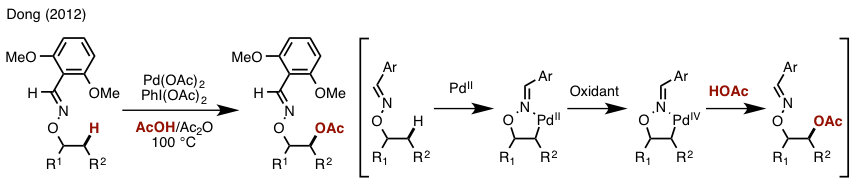
Figure 2. Catalytic β-alcohol sp3 C–H oxidation
Manipulating DG and reaction conditions carefully, they developed sulfornyloxylation instead of acetoxylation. Although sulfornyloxy group is a good leaving group, no elimination occurs because the product does not have acidic a-proton. Finally, treatment with catalytic Pd(OAc)2, oxidant N-fluoroenzenesulfonimide (NFSI), and tosylic acid under 90ºC afforded the corresponding sulfonyloxylation product in 68% yield (Figure 3). It is reported that this reaction is tolerant to moisture and air.
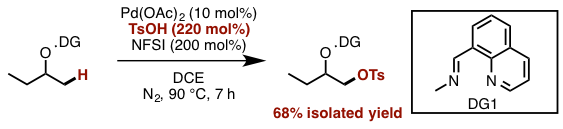
Figure 3. Catalytic β-alcohol sp3 C–H sulfonyloxylation
Application
With optimized conditions in hand, the substrate scope of the reaction was examined. The merit of this reaction is that less-substituted sp3 C–H bonds can be selectively converted to the sulfonyloxy group, and that various functional groups, such as amines and alkenes, can tolerate the reaction condition (Figure 4).
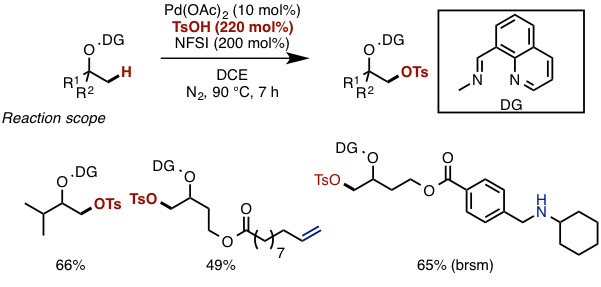
Figure 4. Examination of substrate scope
Furthermore, when the reaction was applied to steroidal alcohol (Figure 5), sulfonyloxylated steroid was successfully obtained in 42% yield over two steps. It is common knowledge that the sulfonyloxy group can undergo various SN2 reactions. Indeed, the authors achieved the rapid synthesis of nine steroid derivatives via SN2 reactions. Directing groups were removed smoothly to afford alcohols with excellent yield under hydrogenation conditions.
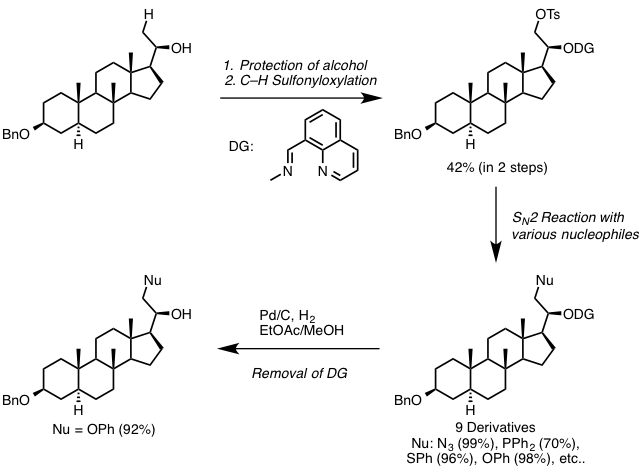
Figure 5. Derivatization of steroidal compounds
Summary
The interesting point of this reaction is that the sulfonyloxy group, which is usually used as a good leaving group, was introduced to the molecule in a nucleophilic fashion. Moreover, the directing group used by the authors not only assists the introduction of sulfonyloxy groups, which can be further converted to various substituents, but also protects alcohols from epoxidation. With their original “tag” and their new method of sp3 C–H functionalization, the repertoire of methods for C–H functionalization has just gotten bigger.
Interview
Finally, we interviewed Mr. Yan Xu, the first author of this paper.
1) Why you get interested in sp3 C–H sulfornyloxylation?
The rapid preparation and intensive testing of a panel of closely related analogues is the basis for modern drug discovery. As a result, strategies allowing for the diverse derivatization of a late-stage intermediate are much more appreciated than the individual parallel syntheses in searching for lead compounds. Since the end of the twentieth century, the activation and functionalization of C–H bonds has emerged as a revolutionary trend for derivatizing complex synthetic intermediates, however, its use for late-stage diversification is still limited, especially at unactivated sp3 positions. Thus, we are quite interested in the development of a new diverse sp3 C−H functionalization strategy through the direct installation of a sulfonyloxy group at an inert C–H bond and the subsequent employment of nucleophilic substitution reactions to prepare various derivatives. We anticipate this method will streamline the preparation of pharmaceutical analogues, expedite the search for lead compounds and ultimately contribute to the discovery of new drugs.
2) The process to achieve the optimal condition
One of the biggest challenges of this chemistry is that we actually cleavage a very inert sp3 C–H bond and replace it with a highly reactive sp3 C–OTs bond. Thus, during the optimization, we balanced carefully between the activity of our catalytic system and the stability of the product under the reaction condition. Luckily, by the evolution of the directing groups and the intensive testing of corresponding reaction parameters (metal pre-catalysts, oxidants, OTs sources, etc), we finally identified the optimal condition for the desired transformation.
3) Future of the related work
One of our ongoing works related to this chemistry is trying to make it more practical. By seeking for more convenient directing group installation/cleavage methods as well as a milder C–H sulfonyloxylation condition. we hope that this strategy will be more appealing and find further use in the drug discovery and natural product synthesis. Besides, given that our directing groups are installed on hydroxyl groups, which are widely found in bioactive molecules and frequently used as synthetic handles, we hope that we could work beyond the activation of the β C–H bonds, and discover some more attractive transformation at the γ or even the δ C–H bonds of alcohols in the future.
Reference
- (a) Giri, R.; Chen, X.; Yu, J.-Q. Angew, Chem., Int. Ed. 2005, 44, 2112. DOI:10.1002/anie.200462884 (b) Rit. R. K.; Yadav. M. R.; Ghosh, K.; Shankar, M.; Sahoo, A. K.Org Lett. 2014, 16, 5258. DOI: 10.1021/ol502337b
- Ren, Z.; Mo, F.; Dong, G. J. Am. Chem. Soc. 2012, 134, 16991. DOI:10.1021/ja3082186
- Desai, L. V.; Hull, K. L.; Sanford, M. S. Am. Chem. Soc. 2004, 126. 9542.DOI: 10.1021/ja046831c
This article written by Japanese is here
Related books
[amazonjs asin=”4759813659″ locale=”US” title=”Fukassei ketsugoÌ”, fukassei bunshi no kasseika : kakushinteki na bunshi henkan hannoÌ” no kaitaku = Bond activation and molecular activation”]